A flexible and biologically informed pipeline that refines highly variable genes from single-cell RNA-seq data to optimize deep learning model training.
1. Introduction
- HVGs are defined as genes that exhibit significantly greater expression variability across single cells than expected by random technical noise, and this variability may reflect underlying regulatory mechanisms or disease-relevant transcriptional dynamics (Hafemeister & Satija, 2019; Yip et al., 2017). In high-dimensional single-cell transcriptomic datasets, however, selecting which of these variable genes are most meaningful as input features for machine learning or deep learning (DL) models poses a significant challenge. Standard approaches typically rely on identifying the top 2,000 to 5,000 HVGs based purely on statistical criteria such as expression variance. These HVGs are often used as the primary input features for training deep learning models in downstream applications such as phenotype prediction, as shown by Martorell-Marugán et al. (2025). However, this selection can include genes that are either noisy, stably expressed housekeeping genes, or biologically irrelevant to the disease or treatment under investigation. For example, ribosomal and mitochondrial genes—although highly expressed and sometimes variable—are often associated with general cellular maintenance rather than condition-specific responses, and may obscure meaningful biological signals [Luecken & Theis, 2019]. Similarly, housekeeping genes, which are broadly and stably expressed across tissues, are unlikely to distinguish between pathological states [Eisenberg & Levanon, 2013]. Even genuinely variable genes may fail to capture condition-specific biological signals if they do not align with meaningful biological processes [Yip et al., 2017]. Therefore, a biologically guided assessment of HVG sets—grounded in curated GO terms or pathway-based references—can significantly improve both the interpretability and effectiveness of model features.
- To address this limitation, we developed a bioinformatics pipeline tailored for single-cell RNA-seq (scRNA-seq) data in 10x Genomics (i.e., genes, barcodes, and matrix files). This pipeline integrates statistical variability with biological relevance by benchmarking HVG sets against a curated list of genes known to be involved in context-specific pathways. In doing so, it helps researchers derive more robust and interpretable gene sets for model training.
- The motivation behind this pipeline stems from the need to better align computational feature selection with biological insight. Instead of relying solely on enrichment analyses post hoc, we proactively incorporate pathway- and GO-term-based biological knowledge derived from literature. This biologically anchored approach increases the interpretability and accuracy of predictive models.
- While the initial use case involved kidney cancer patients treated with immune checkpoint blockade (ICB) therapy, the pipeline is designed to be generalizable. Users can adjust pathway and GO term selections to suit other diseases and treatment modalities.
2. Pipeline Overview
- Step 1: Extract HVGs from selected cell types and generating multiple HVG sets (e.g. 100 to 9,000 genes) for downstream evaluation.
- Step 2: Determine the optimal HVG set by assessing when adding more genes yields minimal gain in overlap with immune-related genes extracted via biomaRt and msigdbr using literature-informed GO terms and pathways.
- Step 3: Generate a refined gene list by intersecting the selected HVG set with immune-related genes derived from literature-informed GO terms and pathways, using biomaRt and msigdbr.
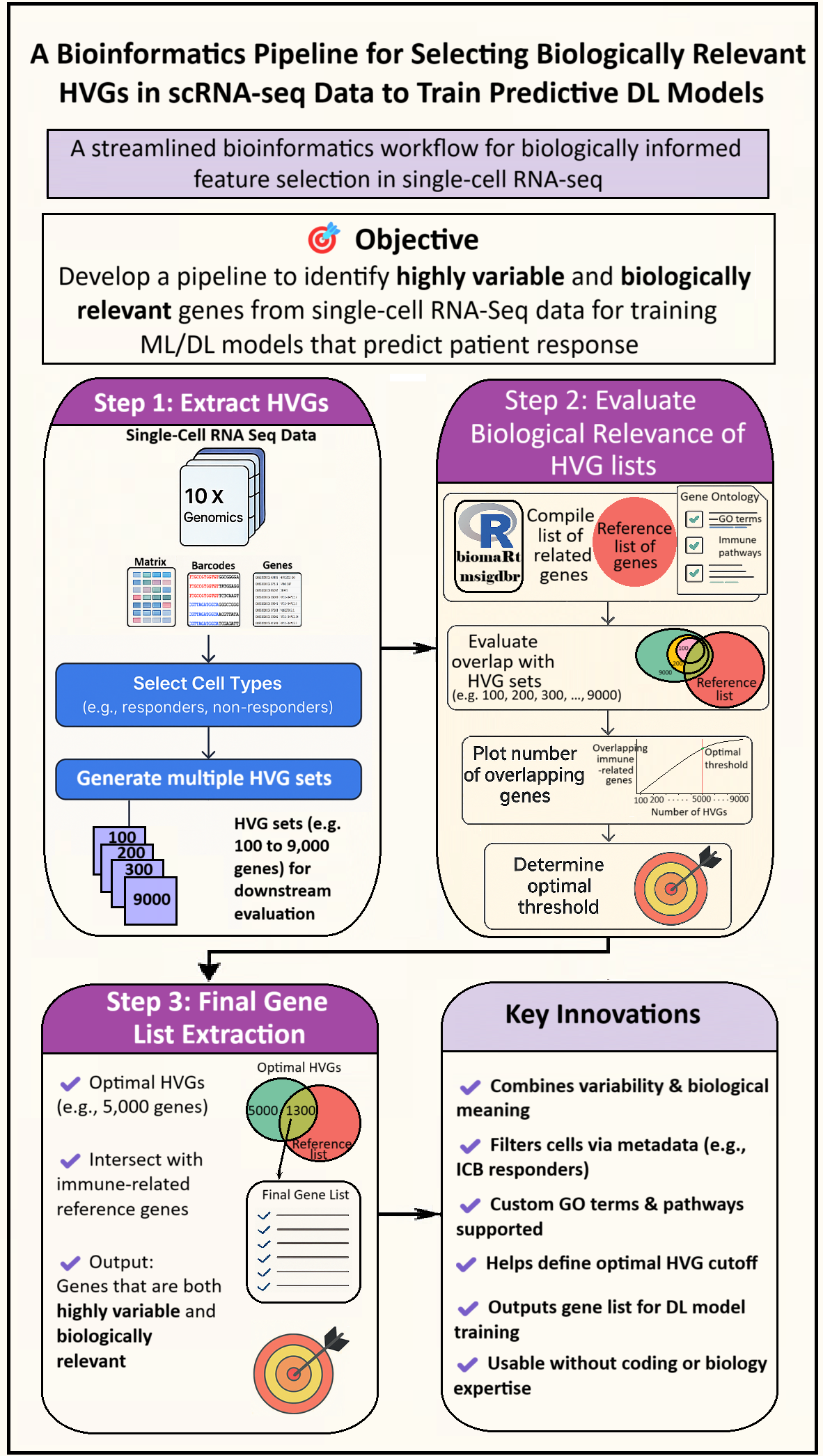
- Step 1: Extract HVGs: Users start by uploading standard scRNA-seq files (10X format) and selecting specific cell types of interest—such as only immune checkpoint blockade (ICB)-exposed cells or responders & non-responders—based on metadata. This tailored inclusion ensures flexibility across research questions. Multiple HVG sets are generated (e.g., 100 to 9,000 genes) to allow evaluation of optimal size later.
- Step 2: Evaluate Biological Relevance of HVG Lists: Using biomaRt and msigdbr packages in R, users compile a custom list of biologically relevant genes based on selected GO terms and immune-related pathways. This list serves as a reference to strong>biologically assess the generated HVG sets. The pipeline visualizes how overlap changes with increasing HVG count and helps pinpoint an optimal HVG threshold that maximizes biological relevance while minimizing redundancy.
- Step 3: Final Gene List Extraction: From the optimal HVG set (e.g., 5,000 genes), the pipeline intersects with the reference gene list to extract a final output: genes that are both highly variable and biologically meaningful - ideal for predictive modeling.
- Key Innovations ✔ Integrates statistical variability with biological meaning ✔ Enables metadata-based filtering (e.g., ICB-Exposed only cells) ✔ Supports user-defined GO terms and pathways ✔ Identifies an optimal HVG cutoff ✔ Generates gene lists for DL/ML model training ✔ Designed for users with minimal coding or biology background & expertise
3. Step-by-Step Breakdown
3.1 Step One: Generate HVG Sets
- The pipeline starts with scRNA-seq input files in 10X format. Users can specify which cell populations to include—for instance, based on therapy response categories such as partial response (PR), stable disease (SD), or progressive disease (PD). The selected cells are saved into a new directory.
- Using Seurat, the pipeline calculates HVGs and saves them incrementally in sets of 100, from 100 up to 9,000. These sets represent candidate feature spaces for downstream evaluation.
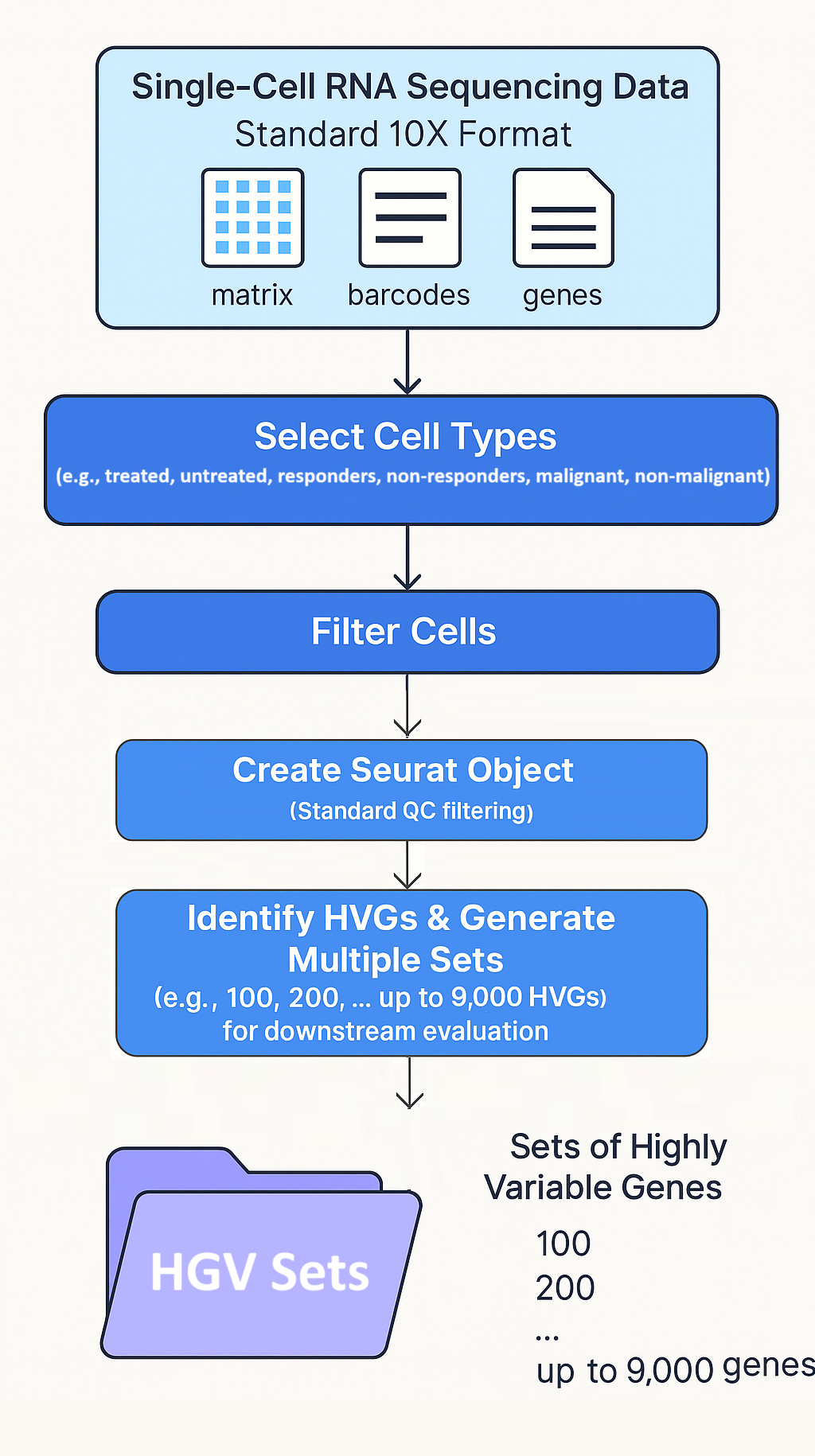
- Figure 3.1. Step One: Generate HVG Sets: In this step, we have preprocessing and HVG Identification Workflow Using Seurat package.
- A stepwise outline of the initial preprocessing phase for single-cell RNA sequencing (scRNA-seq) data using the Seurat package in R. This step forms the foundation of the biologically guided HVG selection pipeline.
- Starting from the standard 10X format (matrix, barcodes, genes), the workflow involves integrating metadata to annotate cells by phenotypes of interest (e.g., treated, untreated, responders, non-responders, malignant, non-malignant). These annotations guide the selection of relevant cell types to be included in the analysis.
- Following cell selection, quality control (QC) filtering is applied during the creation of the Seurat object, including steps such as filtering out low-quality cells based on mitochondrial gene content or gene/cell count thresholds.
- The next step includes data normalization and the identification of highly variable genes (HVGs). Multiple HVG sets are generated by varying the number of top variable genes selected (e.g., 100, 200, … up to 9,000), which are saved for downstream biological relevance evaluation and optimal threshold selection.
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
3.2 Step Two: Assess Biological Relevance of HVG Sets
- In this step, each HVG set is benchmarked against a reference list of biologically meaningful genes. These genes are extracted using the biomaRt and msigdbr R packages, based on a curated set of GO terms and immunotherapy-relevant pathways identified through literature review.
- The pipeline calculates how many genes in each HVG set overlap with this reference list. This allows us to measure not only variability but also biological interpretability. The goal is to find the "elbow point" or inflection threshold where adding more HVGs no longer substantially increases the number of biologically relevant genes captured.
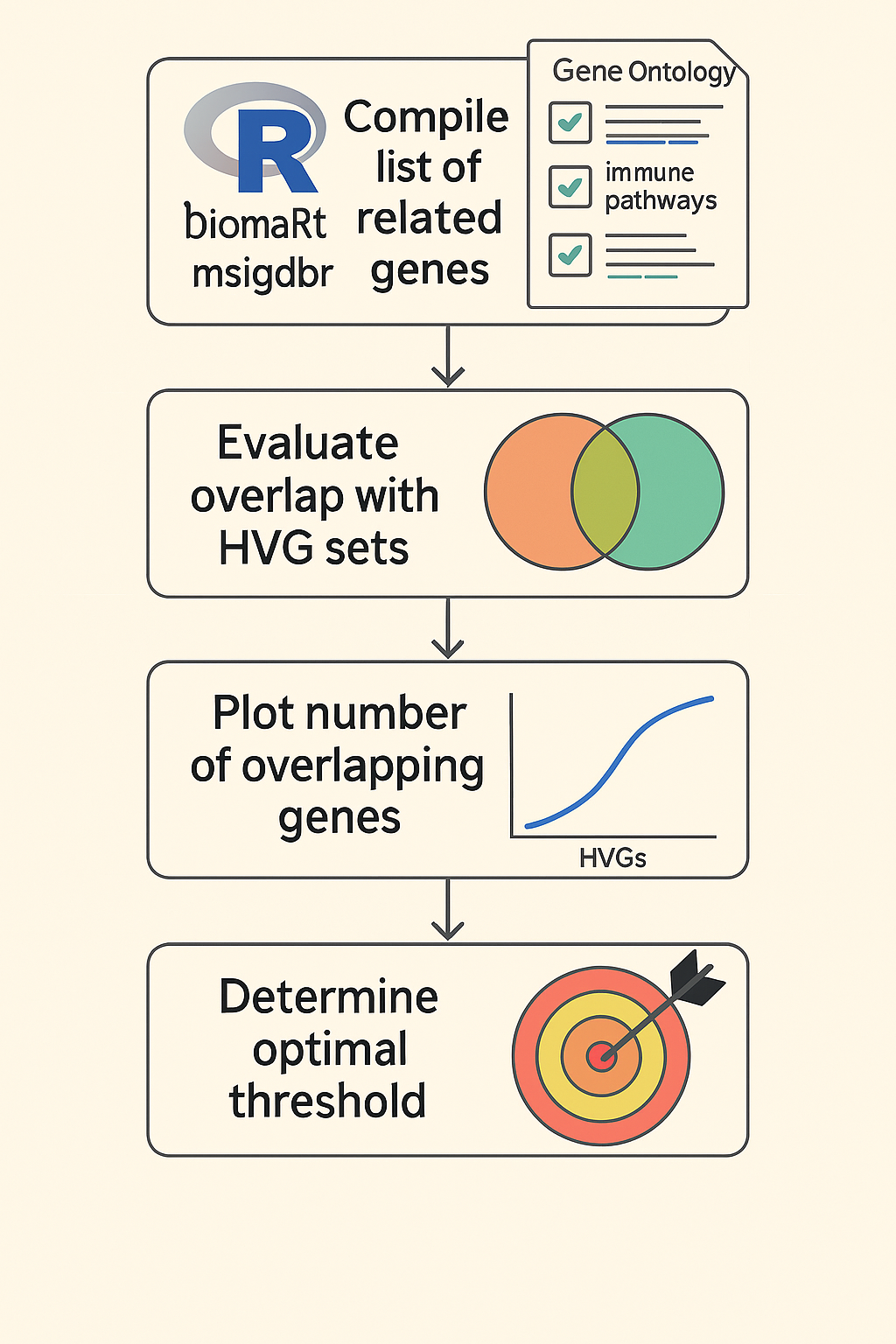
- Figure 3.2. This diagram outlines the process of assessing the biological relevance of each HVG set by benchmarking against curated gene lists (Reference list of genes) (Step 2). The workflow starts with compiling reference gene sets using the biomaRt and msigdbr packages in R, based on Gene Ontology terms and immunotherapy-related pathways derived from literature. Each HVG set is then evaluated for overlap with this reference list, and the number of overlapping genes is quantified and plotted. This visualization helps identify an optimal threshold—typically an inflection point—where increasing the number of HVGs no longer yields substantial gains in biological interpretability.
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
3.3 Step Three: Extract Biologically Relevant HVGs for Model Training
- After identifying the optimal HVG set (e.g., 5,000 genes), the pipeline intersects it once more with the reference gene list. The result is a refined gene set that is both statistically variable and biologically informed, and is now ready to be used as input for deep learning or machine learning models.
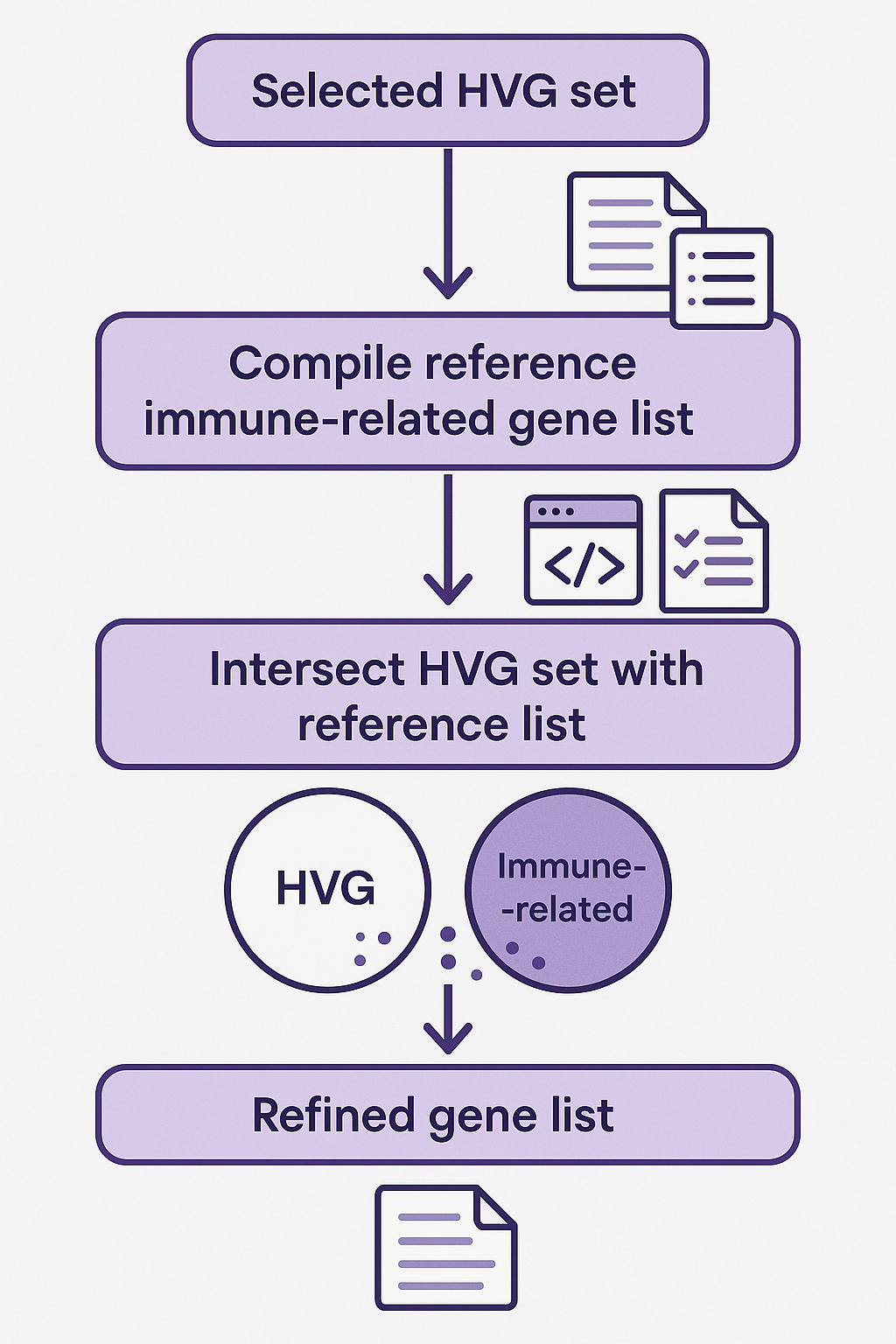
- Figure 3.3. Step Three: This diagram illustrates the final step of the biologically guided HVG selection pipeline. A previously selected HVG set—identified as optimal based on overlap analysis—is intersected with the curated list of immune-related or biologically meaningful genes. This step refines the HVG set, narrowing it down to genes that are both highly variable and functionally relevant. The resulting gene list serves as the final input for downstream machine learning or deep learning model training.
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
# Code will be added here soon
4. Final Output and Applicability
- The final output of the pipeline is a biologically refined list of highly variable genes (HVGs), saved as a .csv file. This gene set serves as a high-quality input for downstream deep learning models aimed at predicting treatment response or other clinically or biologically relevant outcomes — depending on the specific context of the dataset.
- One of the core strengths of the pipeline is its ability to determine an optimal HVG threshold. This threshold reflects the point at which adding more genes no longer significantly increases the number of biologically meaningful genes captured. Selecting fewer HVGs than this optimal point may risk excluding informative genes with lower variability that nonetheless play crucial roles in disease mechanisms or treatment responses. Conversely, selecting too many HVGs may introduce noise by including irrelevant genes, which can negatively impact model training and performance.
- To address this, the pipeline doesn't rely solely on variance rankings. Instead, it incorporates biological filtering using pathway- and GO-term-based reference gene sets relevant to the disease or treatment under investigation. This ensures that even HVGs with lower variability — but biological significance — are retained, while biologically irrelevant genes are excluded. This dual approach improves the interpretability and reliability of feature selection for downstream modeling.